Current Funding
U.S. National Science Foundation
|
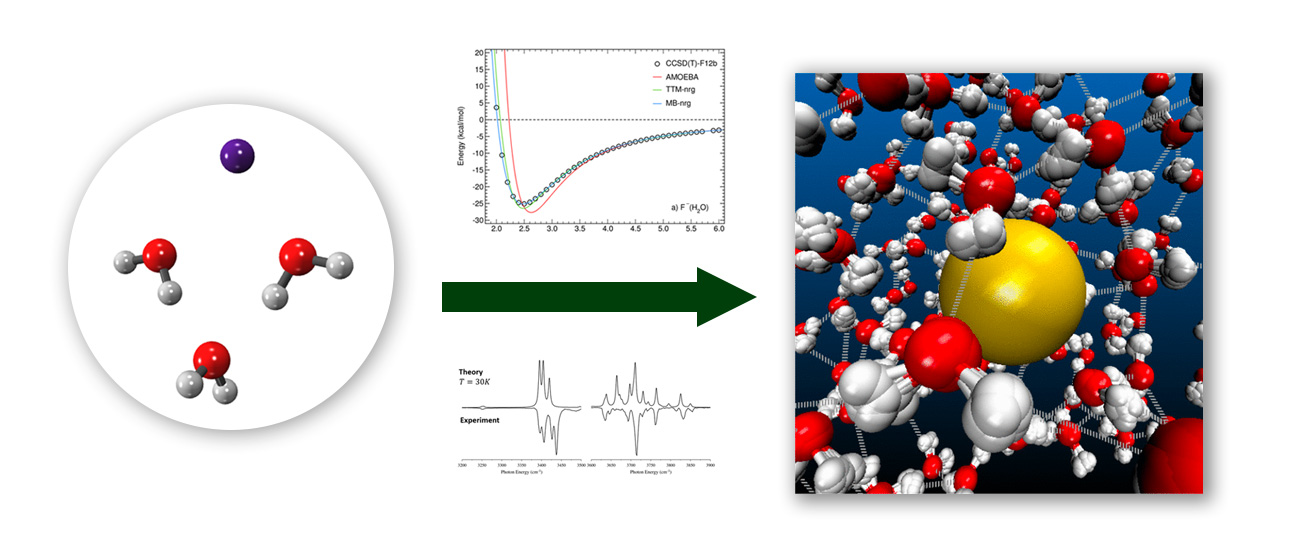 |
Disentangling Many-Body Effects and Couplings in Vibrational Spectra of Aqueous Clusters
The Chemical Structure, Dynamics, and Mechanisms A Program of the NSF Division of
Chemistry funds Professor Francesco Paesani (University of California – San Diego)
and Professor Ryan Steele (The University of Utah) to collaboratively investigate
the manner in which charged particles, called ions, interact with water. This hydration
of ions drives numerous processes, including biological chemistry, atmospheric chemistry,
and renewable-energy chemistry. The response of these chemical species to infrared
light provides clues regarding their behavior, but unraveling the details of this
behavior from experiments often requires large-scale computer simulations. In this
project, the two research groups will use new computer simulations to study, at the
atomic level, the processes that govern these ion-water interactions.
The two research groups will combine newly developed many-body potential energy functions with new local-mode approaches for simulating anharmonic vibrational spectroscopy. This synergy will enable the decomposition of the anharmonic couplings that drive unique vibrational behavior in strong ions and provide insight to recent hydrated-ion experiments. The impact of divalent ions on hydrogen-bonding networks will also be investigated, along with the evolution of hydration behavior of small biomolecular ions. Wherever possible, these investigations will be coupled with recent and forthcoming results from experimental collaborators. Ultimately, these studies will be used to calibrate the potential energy functions and their underlying physics, in order to simulate both small, hydrated ions and condensed- phase solutions.
06/01/2021 - 05/31/2024
$382,527
U.S. Department of Energy
|
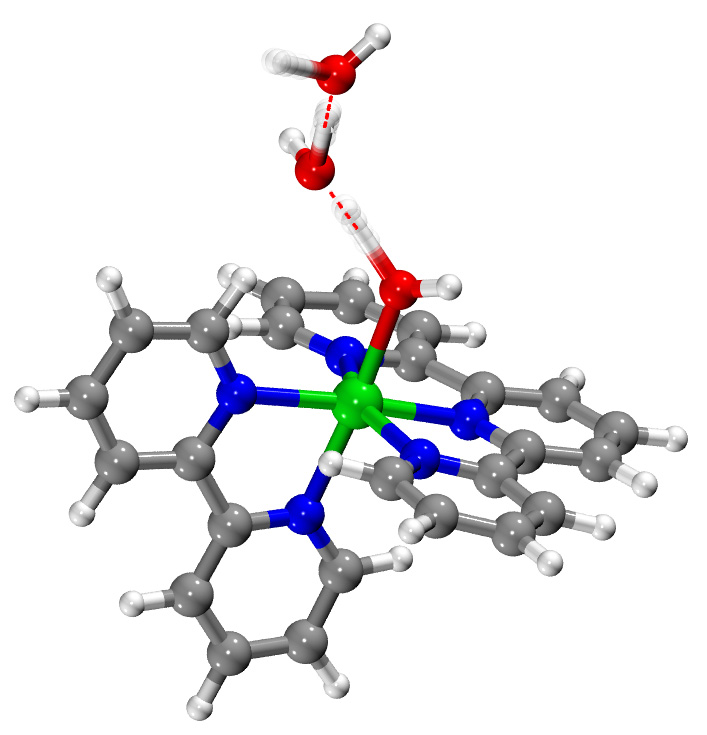 |
Vibrational Signatures of Electronic Properties in Renewable-Energy Catalysis
Solar energy catalysis presently provides as many promises for abundant, renewable
energy as it does questions for the field of chemistry. One of the potential routes
to uncover the molecular-level details of solar energy is vibrational spectroscopy,
which probes the response of molecules to infrared radiation. Considerable progress
has been made along these lines in recent years, but bridging these experiments with
meaningful, chemical insights often requires high-quality computer simulations for
interpretation. This research program will discover and develop new approaches for
computational simulations of this molecular motion. Specifically, the research will
decipher the connection between the behavior of electrons inside of molecules and
the accordant molecular vibrations. Working closely with committed experimental collaborators,
this conceptual and computational framework will be used to explain the results of
new spectroscopy experiments. The resulting products of the research program will
include openly available software and algorithms for the simulation of challenging
vibrational spectra, as well as critical mechanistic insight into energy-focused catalytic
processes that are opaque to other existing analytical techniques.
09/15/2018 - 09/14/2022
$600,000
University of Utah
|
Artificial Intelligence for Molecular Potential Energy Surfaces
The goal of this research program is to develop artificial intelligence platforms
for quantum chemistry computer simulations. Although ab initio computations have become both commonplace and critical to most
research programs, their computational cost makes them inaccessible to many size and
time regimes of Chemistry. Furthermore, the seemingly endless series of "knobs" to be turned in these calculations--with
unknown implications on the resultant accuracy--makes the perpetual cost-vs-accuracy
gamble an impediment to good science.
This work is the beginning of a vision for the retooling of the field of computational chemistry. Rather than recomputing quantum chemistry information anew--with modest levels of accuracy--for each organic molecule (as one example), computing clusters could instead be running in the proverbial background in order to prepare nearly exact calculations on relatively small molecules. From this reference information, machine-learning approaches will be trained to generate quantum chemistry-quality data that can be applied to both molecular and condensed-phase systems. The resulting tools would have quantum chemistry accuracy with only force-field computational cost.
09/1/2021 - 08/31/2022
$48,000
XSEDE
|
Exploring the Interface of Quantum Chemistry and Molecular Motion
The main objective of this research program is to understand the manner in which the electronic structure of molecules impacts molecular motion. This “motion” aspect can be direct, in the sense of pursuing ab initio molecular dynamics simulations of reactive systems, or inferred from vibrational spectral signatures. In all cases, however, this program pursues chemical applications in which quantum chemistry-based simulations are critical, including strong ions, open-shell species, and systems for which spectroscopic accuracy is required. Chemical targets accordingly range from renewable-energy catalysts to biological ions.
09/1/2021 - 08/31/2022
6,770,581 Core Hours
on Expanse (San Diego Supercomputing Center)
Past Funding
U.S. National Science Foundation
Chemical Theory, Models, and Comptuation (CTMC)
New Methods for Dynamical Quantum Chemistry (CAREER)
04/01/2015 - 03/31/2020 (+no-cost extension to 03/31/2021)
$606,515
American Chemical Society
Petroleum Research Fund - New Directions
Metal-H2 Complexes vs Metal Hydrides: Nuclear Motion and Implications for Hydrogenation Catalysis
09/01/2015 - 08/31/2017 (+no-cost extension to 08/31/2018)
$110,000
XSEDE
|
Exploring the Interface of Quantum Chemistry and Molecular Motion
05/13/2013 - 10/01/2014: 50k hour startup
10/01/2014 - 09/30/2015: 3.7 million hours (Value: $263,309)
10/01/2015 - 09/30/2015: 3.5 million hours (Value: $172,228)
10/01/2016 - 09/30/2017: 2.9 million hours (Value: $145,315) + extension
01/01/2018 - 12/31/2018: 1.7 million hours (Value: $ 71,533)
10/01/2019 - 09/30/2020: 3.2 million hours (Value: $ 51,165)
10/01/2021 - 09/30/2022: 6.8 million hours (Value: $ 29,840)
University of Utah
University Seed Grant
*Led to proof-of-concept data for current DOE funding
Chemistry in Action via Anharmonic Spectroscopy
07/01/2013 - 06/30/2014
$28,000